Background
Pyoderma gangrenosum is a rare inflammatory skin condition marked by one or more painful ulcers, often with characteristic violaceous undermined borders accompanied by surrounding erythema, predominantly seen on the anterior lower extremities.1–5 One hallmark finding in ulcerative PG is the pathergy phenomenon, where minor trauma, such as debridement, can either exacerbate an existing ulcer or induce a new one, often leading to a dramatic worsening of the condition.6,7 In addition to its classic ulcerative presentation on the lower extremities, PG also exhibits a bullous form that frequently appears on the face and upper extremities, including the hands.4,5 Clinicians must be aware of the clinical variants of PG, as the diversity of its presentations can often mimic other skin diseases, leading to inaccurate diagnosis and treatment.5,7 Healthcare providers should be vigilant in recognizing potential signs of PG in patients with suspicious lesions and promptly consider appropriate diagnostic testing, screening, and specialist referral.1,5,8
Though the pathogenesis of PG remains unclear, it is described as an inflammatory disease within the spectrum of neutrophilic dermatoses.3–5,9 These dermatoses are characterized by skin lesions that result from a neutrophil-rich inflammatory infiltrate in the absence of infection.1,3,7 Genetically predisposed individuals appear to develop PG.3,5 In this condition, it is proposed that neutrophil dysfunction and an overly active innate immune system trigger the release of pro-inflammatory cytokines and chemokines.3–6 This includes a significant array of interleukins that act synergistically with tumor necrosis factor (TNF)-alpha to activate and amplify the production of matrix metalloproteinases (MMPs). These MMPs, found in high concentrations in PG’s inflammatory infiltration, along with the clonal expansion of T-cells, may play a role in tissue damage and impaired wound healing.3
A substantial portion of PG patients present with concurrent underlying conditions, with inflammatory bowel disease being among the most frequently associated disorders1,2,4–8. IBD arises from a complex interplay of genetic predisposition, immune system dysfunction, environmental triggers, gut microbiota imbalance, and potential autoimmune processes, leading to chronic inflammation and tissue damage in the digestive tract.10 The relationship between IBD and PG is not fully understood, but it is plausible that a connection exists based on shared underlying immune system dysfunction. Given the many uncertainties surrounding PG, case reports such as the one presented here can provide valuable insights into the real-world challenges and nuances of diagnosing and managing this condition.
Case Presentation
A 63-year-old Venezuelan female with a history of hypertension presented to the emergency department due to ten days of worsening swelling to her right face at the angle of the mandible, as well as ulceration and pain with purulent drainage from her right elbow. She also reported concomitant chills, fevers, and severe right side facial pain. The patient added that she had two episodes of bloody stools, the most recent occurring ten days prior to being admitted. Prior to her presentation, the patient received treatment from her primary care physician, including three days of intramuscular Penicillin G benzathine injections and seven days of clindamycin.
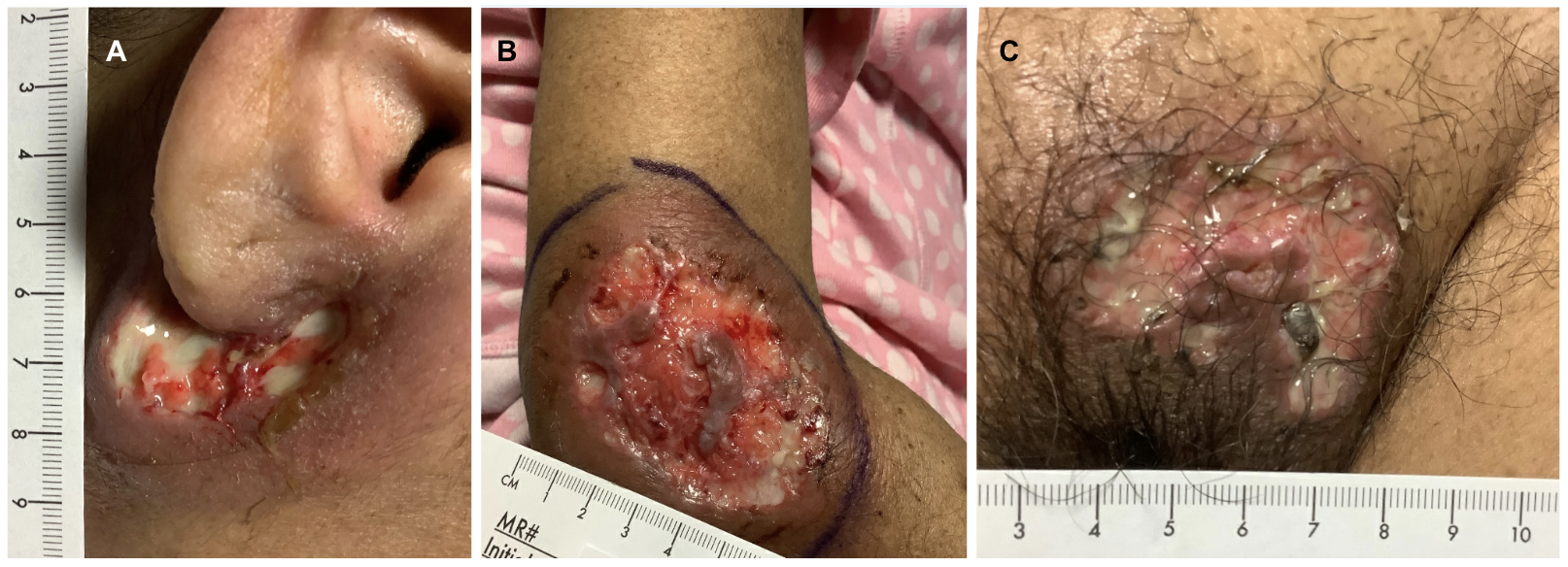
Upon initial evaluation, the patient exhibited mild tachycardia and laboratory results indicated bandemia and hypokalemia. Physical examination revealed purulent drainage from the angle of the right mandible, an ulcerative lesion at the right elbow, and purulent erythema in the pubic region (Figure 1). The patient was admitted for further evaluation of multiple ulcers with suspicion of superimposed bacterial infection and specialists were promptly consulted.

Computed tomography (CT) of the right maxillofacial region revealed cellulitis with phlegmon below the right ear. The underlying right carotid artery appeared isodense and inseparable from inflammatory findings. Magnetic resonance imaging (MRI) of the right elbow showed no evidence of acute osteomyelitis or substantial joint effusion. However, it revealed cellulitis and phlegmon with a soft tissue defect in the olecranon bursa and lateral margins of the elbow. Additionally, mild intramuscular edema of the anconeus muscle was noted. Pelvic MRI showed no evidence of acute osteomyelitis or septic arthritis. All lesions were sampled for culture, fungal, mycobacterium, and pathology ultimately confirmed a diagnosis of PG. The patient underwent a colonoscopy, confirming a diagnosis of IBD.
The patient was initiated on hydrocortisone, and subsequently on infliximab, a TNF-alpha inhibitor, following quantiferon laboratory testing and targeted wound care was implemented. The patient tolerated initial treatment without adverse effects or progression of cutaneous lesions. The patient was discharged on carvedilol, prednisone, pantoprazole, acetaminophen, Florastor probiotics and alprazolam. She was instructed to follow up closely with outpatient dermatology and gastroenterology.
Upon follow-up, the patient’s skin lesions demonstrated progressive improvement, characterized by reduced size, diminished inflammation, and accelerated formation of granulation tissue. Notably, the patient reported complete resolution of pain symptoms with the therapy. Over the course of six months, consistent healing progress was evident through regular assessments, ultimately leading to the complete resolution of the mucopurulent ulcers and the restoration of the skin barrier. The patient continues to receive ongoing care from her gastroenterology specialist, ensuring the maintenance of treatment and necessary adjustments.
Discussion
PG is a neutrophilic dermatosis that typically affects middle-aged adults, with only a few cases occurring per million people annually.5 While the anterior lower leg is the most common location, accounting for 78% of cases in a retrospective review of 103 patients,1 PG can present atypically and mimic other skin diseases. Based on clinical skin findings and concurrent consideration of IBD, pyoderma gangrenosum was included in the differential diagnosis for the patient described above. The right elbow biopsy, which revealed dense neutrophilic infiltrates, further suggested this neutrophilic dermatosis.1,3–5,9
Given the potential relevance of environmental factors, such as differing pathogens or exposures, the patient’s travel history was carefully documented, particularly due to her status as a Hispanic female who had emigrated from Venezuela one year prior to admission. Other potential diagnoses considered included bacterial, fungal, parasitic, and mycobacterial infections, cutaneous vasculitis, malignancy, Behcet’s syndrome, and Sweet’s syndrome.3 Histology ruled out vasculitis and malignancy, while cultures excluded infection. Traditionally, PG was considered a diagnosis of exclusion. However, in 2018, the Delphi consensus7 and the PARACELSUS score9 introduced diagnostic criteria for ulcerative PG, aiming to reduce misdiagnoses and refine patient selection for clinical trials. The patient’s biopsy findings demonstrating a neutrophilic infiltrate, coupled with the exclusion of infection and a confirmed diagnosis of IBD, aligned her presentation with the major and minor criteria of the Delphi consensus for ulcerative PG.7
This case presents a notable manifestation of PG, with mucopurulent ulcers located exclusively in the pubic area and above in a Hispanic female. Early diagnosis of PG is especially important in cases where the condition is associated with underlying systemic diseases. The initial antibiotic treatment, prescribed under the assumption that the lesions were infectious in nature, highlights the need for considering uncommon causes and seeking additional evaluation if standard therapies fail to improve symptoms. Screening for IBD is essential in patients with PG as it is the most commonly reported systemic disease associated with PG,1,2,6 followed by arthritis, hematologic malignancies, and solid malignancies.6 Similarly, PG should be considered in patients with IBD who present with new-onset skin lesions, though both conditions can be diagnosed simultaneously as was seen here. It is important to note that though patients with IBD are about 29 times more likely to develop PG than those without IBD, PG is still rare among IBD patients, affecting only 0.5%.5
Treatment involves a comprehensive approach that often includes wound care, topical therapy, and systemic medications.4 The use of appropriate immunosuppressants, such as infliximab, can help achieve better outcomes and prevent disease progression in patients with concurrent IBD.1,3–5 Drug interactions should be reviewed upon discharge, especially when patients are being treated with systemic agents. Delayed treatment of PG can lead to severe complications such as prolonged antibiotic exposure, unnecessary debridement, delayed wound healing, and disfiguring scarring.3 It is important to note that patients with IBD are predisposed to an increased risk of clostridium difficile infection and are also prone to experiencing worse outcomes, including higher recurrence rates, an elevated need for colectomy, and an increased risk of mortality.11
In conclusion, this case highlights the importance of considering pyoderma gangrenosum, a rare neutrophilic inflammatory disease, in the differential diagnosis of skin ulcers. It underscores the significance of a multidisciplinary approach in diagnosing and managing pyoderma gangrenosum, particularly when the condition presents atypically or in conjunction with other diseases. It’s through such synergetic efforts that patients receive a comprehensive evaluation to help ensure accurate diagnosis, optimal treatment planning, and timely intervention to prevent complications and improve patient outcomes. The case also provides valuable insights into the potential for atypical presentations and emphasizes the importance of exploring potential systemic associations. Despite the passage of nearly a century since its description, the absence of a definitive ‘gold standard’ therapy persists on account of the intricate interplay of immune dysregulation, scarcity of clinical trial data, and diverse treatment responses. The call for further research focusing on elucidating the underlying mechanisms and evaluating targeted therapeutic interventions remains integral to advancing our understanding and optimizing patient care.
Author Contributions
All authors have reviewed the final manuscript prior to submission. All the authors have contributed significantly to the manuscript, per the International Committee of Medical Journal Editors criteria of authorship.
-
Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND
-
Drafting the work or revising it critically for important intellectual content; AND
-
Final approval of the version to be published; AND
-
Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of Interest/Disclosures
The authors have no conflicts of interest to disclose.
Corresponding Author
Lorena P Bonilla, MD
Herbert Wertheim College of Medicine, Florida International University
Baptist Health Medical Group, Baptist Health South Florida
Email: bonill07@gmail.com